Autoimmunity
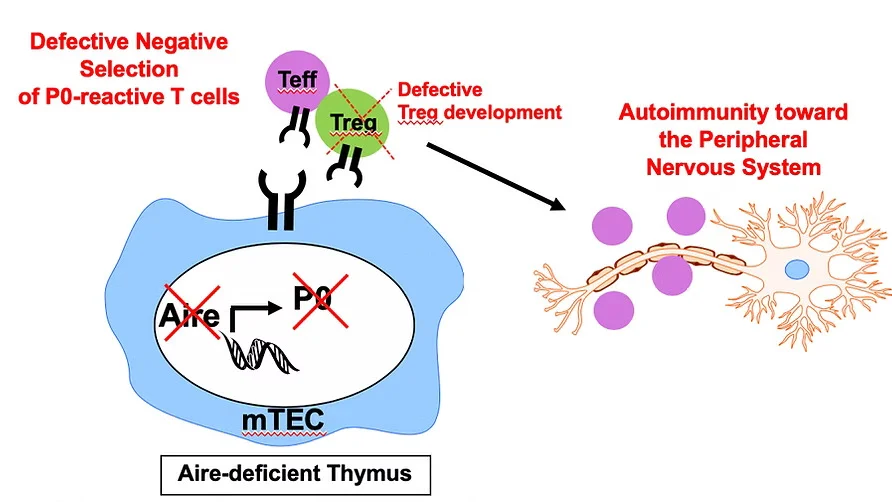
Defective Aire-dependent central tolerance to myelin protein zero is linked to autoimmune peripheral neuropathy
Aire deficiency predisposes to peripheral nervous system (PNS) autoimmunity. We identified myelin protein zero (MPZ or P0) as a PNS-specific Aire-regulated antigen expressed in the thymus. Aire deficiency allows escape of P0-specific T cells to escape negative selection to cause PNS autoimmunity. We discovered that autoimmune-prone NOD mice with a dominant Aire G228W mutation develop spontaneous PNS autoimmunity that resembles features of Guillain-Barré Syndrome and its chronic counterpart, chronic inflammatory demyelinating polyneuropathy (CIDP) (Su et al, J Immunol, 2012). APS1 patients have also been reported to develop CIDP, suggesting that Aire deficiency also predisposes to PNS autoimmunity in humans. See our review on CIDP pathogenesis: Wolbert et al, JCI Insight, 2020
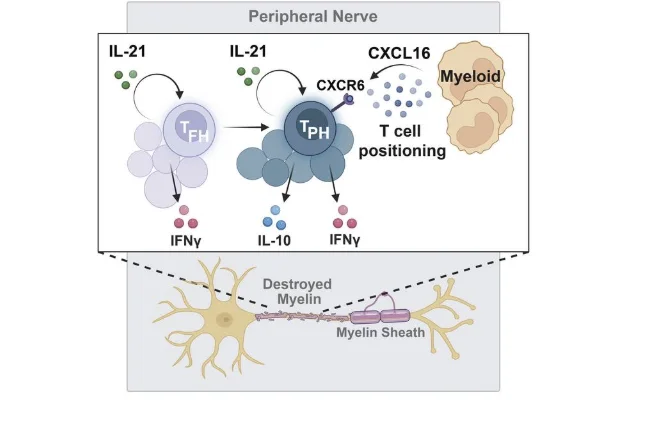
A pathologically expanded, clonal lineage of IL-21–producing CD4+ T cells drives inflammatory neuropathy
Among T cells, CD4+ T cells in particular are critical in the pathogenesis of inflammatory neuropathies. CD4+ T cells are increased in peripheral nerves of patients with inflammatory neuropathies and SAPP mouse models, suggesting a role for CD4+ T cells in peripheral nerve myelin destruction. We showed that, in peripheral nerve infiltrates of neuropathic Aire-deficient NOD mice, terminally differentiated effector CD4+ T cells were clonally expanded and expressed IL-21. These IL-21–producing cells could be grouped into 2 transcriptionally distinct populations, which resembled T follicular helper (Tfh) and T peripheral helper (Tph) cells. Notably, TCR clonotypes were shared in these 2 subsets, supporting the idea of a common lineage for these 2 cell populations. Additionally, we demonstrate that IL-21 signaling was required for neuropathy development and that IL-21 upregulated CXCR6, a chemokine that promotes CD4+ T cell localization within peripheral nerves. Read the paper: Seyedsadr et al, JCI, 2024
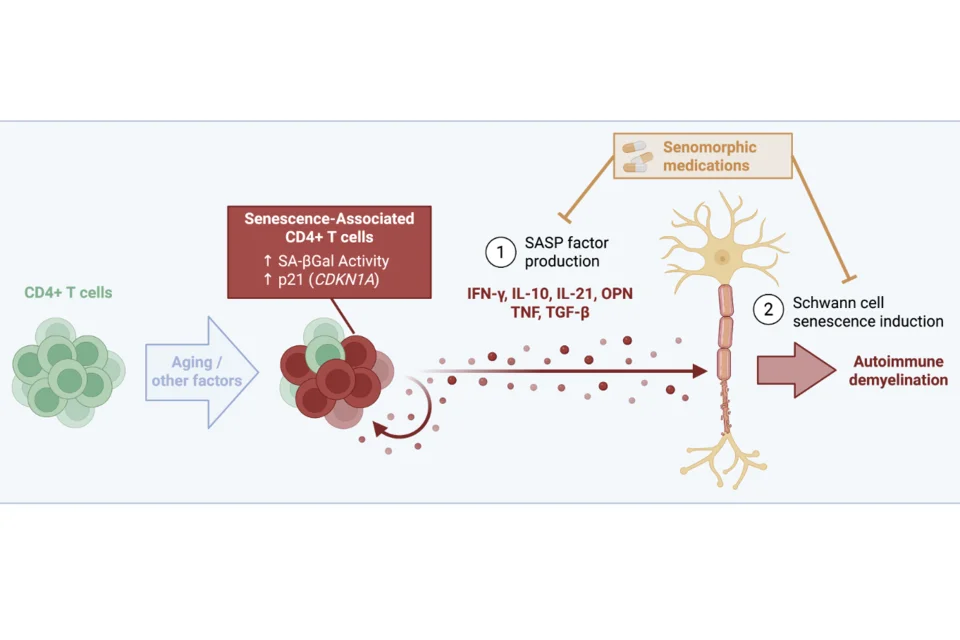
Pathologic T cell immunosenescence drives the development of age-associated autoimmune peripheral neuropathy
While certain autoimmune conditions occur commonly in the young, others are more frequent in the aged. A striking example is chronic inflammatory demyelinating polyneuropathy (CIDP), an autoimmune disease of peripheral nerves that occurs at a peak decade of onset of 70-79 years. How aging predisposes to autoimmunity, however, remains unclear. In CIDP patients, we identified an expanded population of T cells that exhibit hallmark senescence features, including increased SA-βGal activity and higher CDKN1A expression. These senescence associated T cells express multiple senescence associated secretory phenotype (SASP) factors (IFN-γ, TNF-α, TGF-β, IL21, Spp1) and demonstrated an enhanced capacity for inciting neuropathy in a CIDP mouse model. Notably, SASP suppression by a clinically available senomorphic therapy dampened senescence features in peripheral nerves and protected mice against neuropathy. Together, these findings delineate a key role for T cells exhibiting a pro-inflammatory SASP in predisposing to age-associated autoimmune disease. Read the preprint: McCarthy et al, Biorxiv, 2026
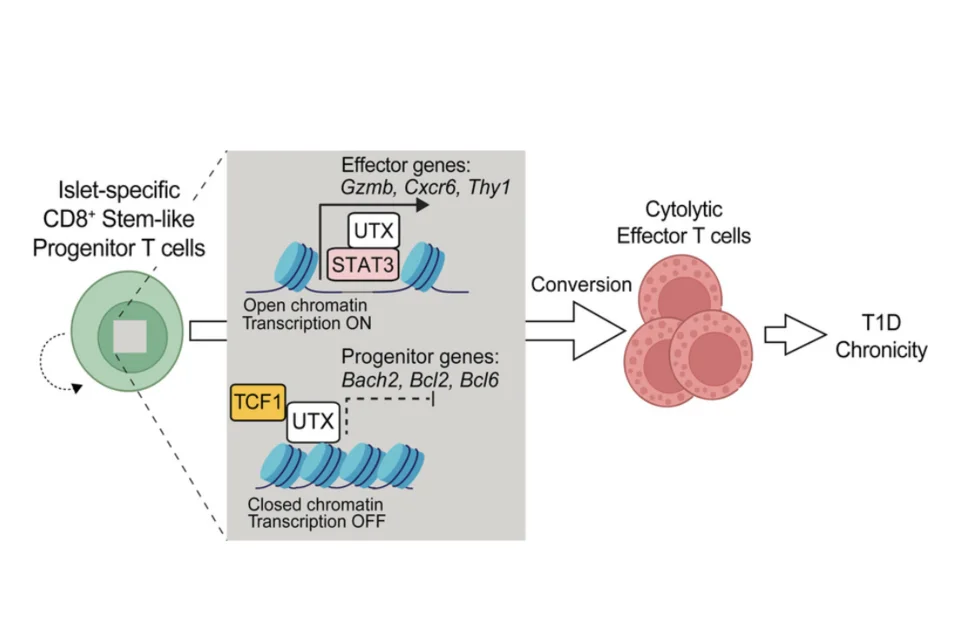
UTX coordinates TCF1 and STAT3 to control progenitor CD8+ T cell fate in autoimmune diabetes
Type 1 diabetes mellitus (T1D) is a chronic disease caused by an unremitting autoimmune attack on pancreatic β cells. This autoimmune chronicity is mediated by stem-like progenitor CD8+ T cells that continually repopulate the pool of β cell–specific cytolytic effectors. Factors governing the conversion of progenitors to effectors, however, remain unclear. T1D has been linked to a chromosomal region (Xp13-p11) that contains the epigenetic regulator UTX, which suggests a key role for UTX in T1D pathogenesis. Here, we show that T cell–specific UTX deletion in NOD mice protects against T1D development. In T cells of NOD mice and patients with T1D, UTX ablation resulted in the accumulation of CD8+ progenitor cells with a concomitant decrease of effector cells, suggesting a key role for UTX in poising progenitors for transition to effectors. Mechanistically, UTX’s role in T1D was independent of its inherent histone demethylase activity but instead relied on binding with transcription factors (TCF1 and STAT3) to coregulate genes important in the maintenance and differentiation of progenitor CD8+ T cells. Read the paper: Chen et al, JCI, 2025